
AlphaFold3: From Protein Structures to Biomolecular Interactions — and What Comes Next
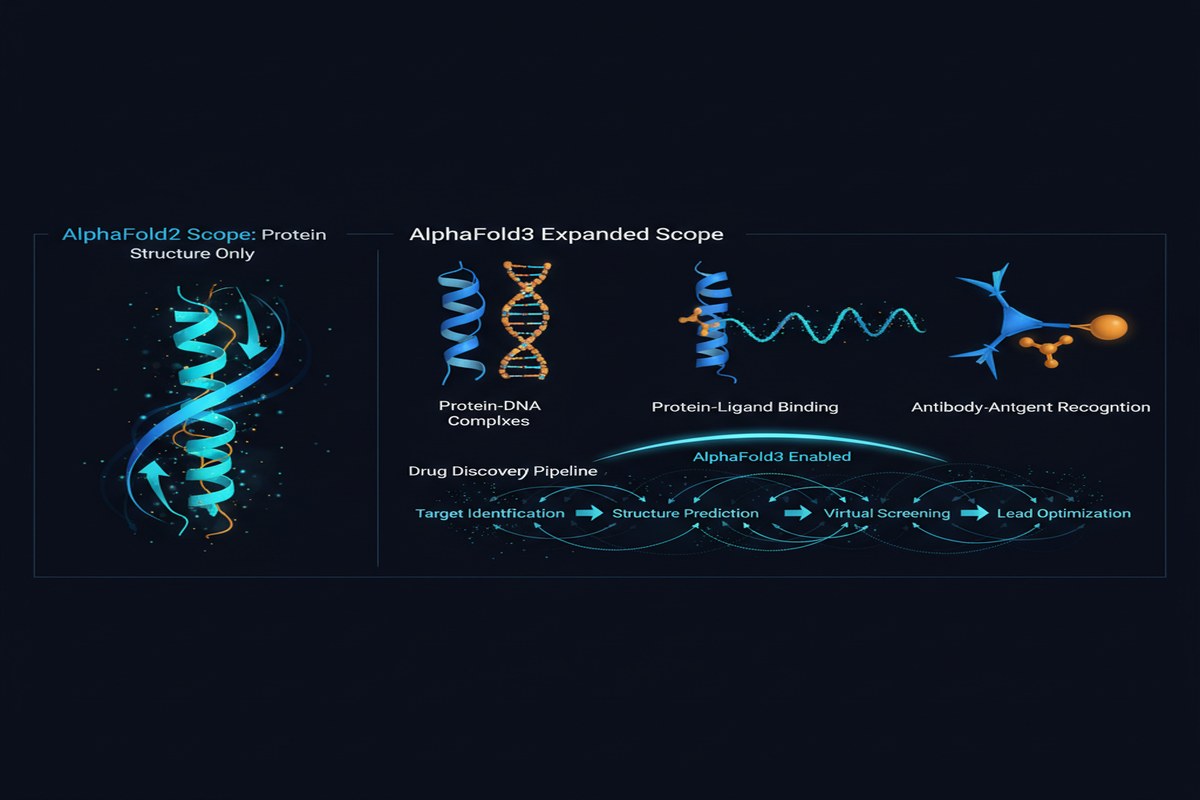
AlphaFold2 solved protein structure prediction. Not approximately solved — comprehensively solved, to an extent that rendered the CASP competition (the protein structure prediction benchmark, once called "biology's space race") nearly moot for single proteins.
The follow-up question was always: what about everything else? Proteins don't work in isolation. They bind to small molecules (drug candidates). They interact with DNA and RNA. They form complexes with other proteins. Understanding these interactions is where the real applications live — drug design, disease understanding, therapeutic development.
AlphaFold3 (Nature, May 2024; comprehensive benchmarking published 2025) is Google DeepMind's answer to this question. It extends AlphaFold's prediction capabilities to virtually all major biomolecular interaction types through a fundamentally different architecture: a diffusion model rather than AlphaFold2's structure module.
What AlphaFold3 Predicts That AlphaFold2 Couldn't
AlphaFold2 predicted single protein structures from amino acid sequences with extraordinary accuracy. AlphaFold3 predicts:
graph TD
AF3[AlphaFold3\nDiffusion-Based Structure Prediction]
AF3 --> A[Protein-Protein\nInteractions]
AF3 --> B[Protein-Ligand\nDocking\nSmall molecule binding]
AF3 --> C[Protein-DNA\nComplexes]
AF3 --> D[Protein-RNA\nComplexes]
AF3 --> E[Single Proteins\nWith modifications]
AF3 --> F[Modified residues\nGlycoproteins\nMetalloenzymes]
A --> G[Drug Target\nDisease Biology]
B --> H[Drug Discovery\nLead Optimization]
C --> I[Gene Regulation\nCRISPR]
D --> J[RNA Processing\nSplicing]
style AF3 fill:#2563eb,color:#fff
style B fill:#059669,color:#fff
style H fill:#059669,color:#fff
The protein-ligand docking capability is the most commercially significant. Drug discovery depends critically on understanding how candidate small molecules bind to target proteins. Accurate binding pose prediction tells you whether a drug candidate will interact with its target in a useful way.
Traditional computational docking (AutoDock, Glide) uses physics-based scoring functions. They're reasonably fast but limited in accuracy. AlphaFold3's learned diffusion-based approach shows substantially improved accuracy on bound protein-ligand complexes.
The Architecture: Why Diffusion?
AlphaFold2's architecture used transformer layers followed by a specialized "structure module" that explicitly modeled the geometry of protein chains. This worked brilliantly for proteins but didn't generalize cleanly to small molecules, nucleic acids, and arbitrary molecular complexes.
AlphaFold3 replaces the structure module with a diffusion model — the same family of models that powers image generation (Stable Diffusion, DALL-E). Instead of explicitly modeling chain geometry, it learns to generate 3D atomic coordinates through a diffusion/denoising process.
flowchart LR
A[Sequence + Structural\nInputs] --> B[AlphaFold3\nInitial Representation]
B --> C[Noise Schedule]
C --> D[Noisy 3D Coordinates]
D --> E[Diffusion Model\nDenoising Steps]
E --> F[Predicted 3D Structure\nAll atom positions]
style E fill:#7c3aed,color:#fff
style F fill:#059669,color:#fff
The diffusion approach handles the heterogeneity of molecular systems naturally — it doesn't need special cases for proteins vs. ligands vs. nucleic acids. Everything is just atoms in 3D space, and the diffusion model learns to generate their positions from the sequence and chemical information.
What the Comprehensive Benchmarking Reveals
A 2025 PMC benchmarking study evaluated AlphaFold3 systematically across all its prediction categories. The results are nuanced:
Strengths:
- Protein-protein interaction prediction: significantly better than prior methods
- Antibody-antigen complex prediction: substantial improvement over alternatives
- Protein-ligand binding pose prediction: best-in-class for most common drug-like molecules
Limitations:
- Static structure prediction: AlphaFold3 predicts a single structure. Real proteins are dynamic — they adopt multiple conformations, and the distribution of conformations matters for function. AlphaFold3 predicts one state "considerably better than AF2" but still with "significant room for improvement" on alternative conformations.
- CASP16 performance: Despite the fanfare, AlphaFold3 did not rank first at CASP16. For some complex tasks, the top human-guided methods still outperform AF3.
- Intrinsically disordered regions: Proteins that don't fold into stable structures are predicted poorly by all structure prediction methods, including AF3. Many disease-relevant proteins (oncoproteins, regulatory proteins) have large disordered regions.
- Novel binding modes: For ligands that bind through mechanisms not well-represented in training data, AF3's performance drops substantially.
The Drug Discovery Applications
The pharmaceutical industry has moved faster than most expected on AlphaFold integration. As of 2025:
- The AlphaFold Protein Structure Database (updated to align with UniProt 2025_03) contains structures for virtually all known proteins
- Multiple drug programs have used AlphaFold3 for binding site identification and lead compound optimization
- Several biotech companies have been founded specifically to exploit AlphaFold-enabled drug design
The most straightforward application: structure-based drug design for targets whose experimental structures are unavailable. AlphaFold3 provides predicted structures that can seed virtual screening and docking calculations.
A more ambitious application: rational protein design. If you can predict how proteins interact with small molecules, you can design proteins that bind desired molecules — creating biosensors, therapeutic proteins, or enzyme catalysts.
SiteAF3 (PNAS, 2026) extends this further: a method for accurate site-specific folding via conditional diffusion built on AF3. By fixing the receptor structure and incorporating binding pocket information as a condition, SiteAF3 consistently outperforms AF3 on complex structure prediction for known binding sites — important for structure-based drug design where the binding pocket location is known.
What's Next for Structural Biology AI
The AlphaFold3 paper in the Nature Nobel Prize context (John Jumper and Demis Hassabis won the 2024 Chemistry Nobel for AlphaFold) frames this as the beginning of a new era, not its completion.
Immediate frontiers:
- Conformational ensemble prediction: Predicting the full distribution of structures a protein adopts, not just one
- Dynamics simulation: Integrating structure prediction with molecular dynamics to understand protein motion
- De novo protein design: Using diffusion models to design proteins with desired structures and functions (RFdiffusion, ProteinMPNN already do this)
- Drug-induced conformational changes: Predicting how a bound drug changes the protein's structure
Longer-term frontiers:
- Whole-cell structural biology: Predicting the structures and interactions of all proteins in a cell simultaneously
- Disease mechanism modeling: Understanding how disease-causing mutations affect protein structure and interactions
- Personalized medicine: Modeling structural effects of patient-specific mutations on drug binding
Why This Matters Beyond Biology
AlphaFold's impact extends beyond protein biology because it demonstrates a general principle: learning from massive datasets of physical structures can solve longstanding prediction problems in natural science.
This template is being applied to:
- Crystal structure prediction (AlphaCrystal, GNOME)
- RNA structure prediction (EternaFold, Ufold)
- Molecular property prediction for materials science (materials informatics)
- Climate and weather prediction (GraphCast, Pangu-Weather)
The methodology — deep learning + physical symmetry constraints + massive training data — is a recipe that generalizes across physical science prediction problems.
My Take
AlphaFold3 is extraordinary. The extension from single proteins to the full universe of biomolecular interactions through a unified diffusion architecture is a genuine scientific achievement. The drug discovery implications are real and significant.
But I want to push back on the narrative that AI has "solved" structural biology. The conformational ensemble problem — predicting not just one structure but the ensemble of structures a protein adopts — remains largely unsolved. For understanding protein function, allosteric regulation, and drug mechanisms, the ensemble matters enormously. AlphaFold3 gives you the single most likely structure; reality gives you a distribution.
The CASP16 result is also worth noting: AlphaFold3 didn't win. For the most complex prediction tasks, expert-guided methods still compete effectively. This suggests there are still insights human expertise provides that the model hasn't fully captured.
The 2025 Nobel recognition validated the AlphaFold work appropriately. Now the task is pushing beyond it — especially toward dynamics, conformational heterogeneity, and true protein design (not just structure prediction). The tools are more powerful than ever. The hard problems remain.
Paper: AlphaFold3 (Nature, May 2024). Benchmarking: "A Comprehensive Benchmarking of AlphaFold3 for Predicting Biomacromolecules and Their Interactions" (2025). Database update: "AlphaFold Protein Structure Database 2025" (Nucleic Acids Research, 2025).
Explore more from Dr. Jyothi


